
















 59 / 74
59 / 74

CARDIOVASCULAR JOURNAL OF AFRICA • Volume 29, No 6, November/December 2018
AFRICA
391
most likely on the basis of increased foetal cardiac afterload. The
combined phenotypes of PE and IUGR, however, worsen foetal
myocardial cardiac function.
51
Tracking MPI measurements may therefore allow the clinician
to monitor myocardial performance of foetuses in severe early-
onset PE across deteriorating placental vascular resistances.
In our opinion, the best way of monitoring the foetus in PE
would be the development of a combined model, consisting
of parameters assessing cerebral circulation as reflected in
the MCA Doppler, placental circulation as epitomised in UA
Doppler, cerebro-placental ratio (combining the previous two
parameters as a ratio), foetal cardiac function, as reflected in
MPI and E/A ratio, placental biomarkers, i.e. sFlt-1 and PLGF,
and maternal biochemistry assessing uric acid and platelet
levels and liver function (representing anti-angiogenic effects of
placental biomarkers in the maternal circulation).
This combined model represents different aspects of the multi-
organ nature of the PE syndrome, taking into consideration the
foetal, placental and maternal units as a continuum, as these
three components are intrinsically intertwined in the disease
process, and would be close to giving a proper and scientific
holistic overview of the foetal condition at a specific time. Using
this combined model would greatly assist the clinician in timeous
delivery of the foetus in a pre-eclamptic scenario. This combined
model represents the culmination of 20 years of international
research in this field.
44-48
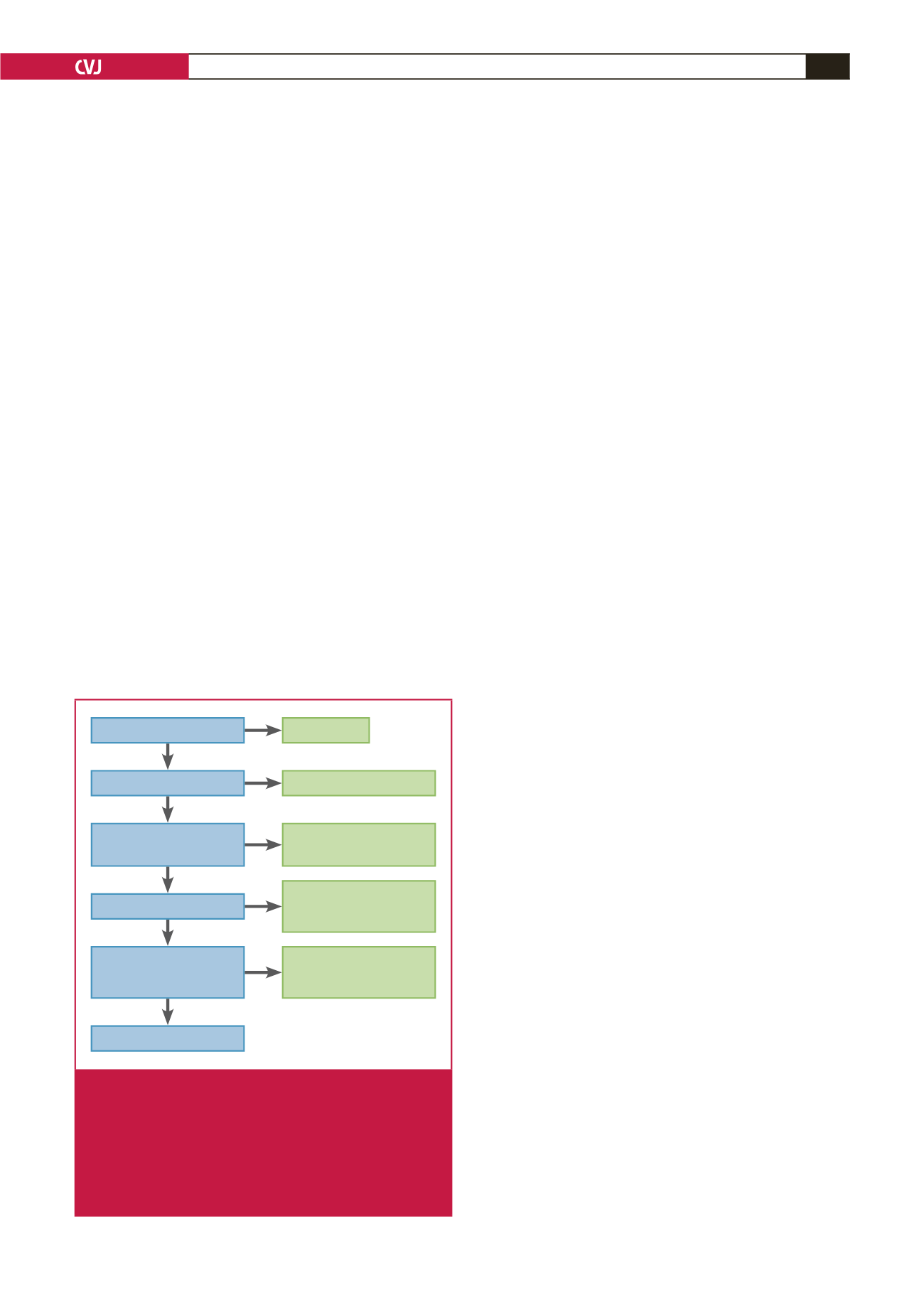
A clinical algorithm using sonography and Doppler to track
the cascade of cardiovascular deterioration of the foetus in PE
is presented in Fig. 1.
Late-onset PE: relative utero-placental ischaemia
Most of our prediction models, as described above, and
management are generally reserved for early-onset PE. Late-
onset PE (
>
34 weeks) accounts for the vast majority of
pre-eclamptic cases. However absolute utero-placental ischaemia
appears to be less relevant in the pathogenesis of late-onset
compared to early-onset PE.
20
We also know that half of patients with late-onset PE do
not have placental histological lesions consistent with maternal
under-perfusion,
52
and that late-onset PE is frequently associated
with foetuses that are appropriate or large for gestational age.
It is possible that in these cases, increased foetal demand for
substrates that surpasses the placental ability to sustain foetal
growth may induce foetal signalling for placental overproduction
of anti-angiogenic factors, and subsequently, compensatory
maternal hypertension.
Late-onset PE may therefore be due to a mismatch between
limited utero-placental blood flow and increased foetal demand
for nutrients, resulting in relative utero-placental ischaemia,
and this could be central to its development. Therefore it is
not surprising that prediction of late-onset PE using first-
and second-trimester biochemical or biophysical (UtADV)
parameters or a combination of these has been less effective
than the prediction of early-onset PE.
20
The reasoning is that it is
probably because a trophoblastic ischaemic threshold leading to
pre-eclampsia is crossed late in pregnancy or due to a more acute
nature of the insults to foetal supply line.
The concept of foetal signalling is central to the pathogenesis
of late-onset PE. The possible pathogenesis described for late-
onset PE has implications, revealing difficulties in its prediction
and the assessment of foetal compromise, as most of the
conventional antenatal surveillance techniques described above
to predict foetal compromise break down. On the other hand, the
late-onset nature of the disease process has far less implications
for prematurity and may therefore not present that much of
a clinical conundrum as far as management is concerned,
compared to the preterm severely pre-eclamptic foetus.
Late-onset pre-eclampsia may be due to a mismatch between
limited utero-placental blood flow and increased foetal demand
for nutrients, resulting in relative utero-placental ischaemia, with
the trophoblast ischaemic threshold crossed late in pregnancy,
and this could be central to its development.
Prophylaxis of PE
Two large meta-analyses
53,54
have suggested that the prophylactic
use of low-dose aspirin is associated with a significant decrease
in perinatal death associated with PE, IUGR and preterm birth,
provided the treatment is initiated before 16 weeks’ gestation.
These meta-analyses reveal a reduction of almost 50% in severe
PE if the aspirin is commenced early (
<
16 weeks). In an article
by Duley
et al
.,
55
low-dose aspirin was associated with an 18%
reduction in foetal and neonatal death in women recruited before
20 weeks’ gestation. The 16-week cut-off point for these studies
is significant because placental implantation and transformation
of uterine spiral arteries are mostly complete by 16 to 20 weeks’
gestation.
56
More specifically, histological studies suggest that
endovascular trophoblastic invasion of uterine spiral arterioles
starts around eight to 10 weeks, and the later myometrial phase
starts around 14 to 15 weeks and completes by 16 weeks.
56
The
SGA
AC or EWF < p10 for GA
Umbilical artery PI > p95
Middle cerebral artery PI
< p5
Ductus venosus PI > p95
MPI > 0.7/absent A wave
in DV/ reversed flow in UA/
abnormal CTG/poor BPP
Foetal death
Uncompensated IUGR
Compensated IUGR
→
onset of hypoxia
? severe hypoxia/early
acidosis
→
early critical
status IUGR
Late critical status
IUGR (high suspicion of
acidosis)
MPI tracking
Fig. 1.
A clinical algorithm using sonography and Doppler to
track the cascade of cardiovascular deterioration of
the foetus in pre-eclampsia. AC = abdominal circum-
ference, EWF = expected foetal weight, IUGR = intra-
uterine growth restriction, SGA = small for gestational
age, DV = ductus venosus, CTG = cardiotocography,
BPP = biophysical profile, PI = pulsatility index, MPI =
myocardial performance index.