


















 57 / 70
57 / 70

CARDIOVASCULAR JOURNAL OF AFRICA • Volume 30, No 5, September/October 2019
AFRICA
299
resolution melting has significantly improved the investigation
for mutations.
The patient population meeting the phenotypic criteria for
FH comprised indigenous Africans (< 1%), subjects of Indian
ancestry (1%) and mixed ancestry (45%), and whites (53%).
In all, 2 200 patients with FH have been genotyped to detect
mutations in exons 4, 7, 8 and 9 of the LDL receptor. The
proportion of FH patients in whom pathogenic mutations were
identified is 57%, of which 96% was in the LDL receptor, 3%
in apolipoprotein B and 1% in PCSK9. In all there were 87
mutations in the LDL receptor gene, four in apolipoprotein B
and four in PCSK9 but some novel mutations have not been
resolved. The 10 commonest mutations in the LDL receptor
were in exons 4, 7, 8 and 9: D206E, V408M, D154N, D200G,
del197, G361V, C356Y, R329X and F382S, and a splice-site
mutation at c.941-4G
>
A.
Several observations were made about possible founder or
regional predelictions for mutations. In the Afrikaner, additional
mutations were identified beyond the original three mutations.
Certain mutations predominated in persons of Jewish and
Indian (Gujerat) origin. The six commonest mutations in the
LDL receptor accounted for
>
90% of the first 10 mutations
(Table 1), and below this the numbers are low for each of the
remaining mutations (< 1% of cohort of identified genotypes).
The importance of recognition of the FH phenotype was
stressed as this has a high and remediable risk of coronary
artery disease with a special need for testing the family owing
to the dominant inheritance of the monogenic causes. A genetic
diagnosis is vital in certain settings, such as in counselling
heritability in pregnancy planning and borderline cases of
hypercholesterolaemia. Genotype–phenotype correlations are of
interest as well as genes aggravating or ameliorating the outcome.
Special clinics for clinical and laboratory evaluation are important
until lipidological skills are improved at undergraduate and
44 years
Hypercholersterolaemia
LDL-C 3.7 mmol/l
50 years
Hypercholersterolaemia
LDL-C 6.2 mmol/l
SNV score 1.002
80 years
Hypercholersterolaemia
LDL-C 3.5 mmol/l
SNV score 0.684
Stroke, CABG
79 years
Hypercholersterolaemia
LDL-C 4.2 mmol/l
SNV score 0.780
40 years
Hypercholersterolaemia
LDL-C 4.4 mmol/l
SNV score 0.931
70 years
Myocardial infarction, 45 years
Myocardial infarction
2 of 7 siblings, MI < 60 years
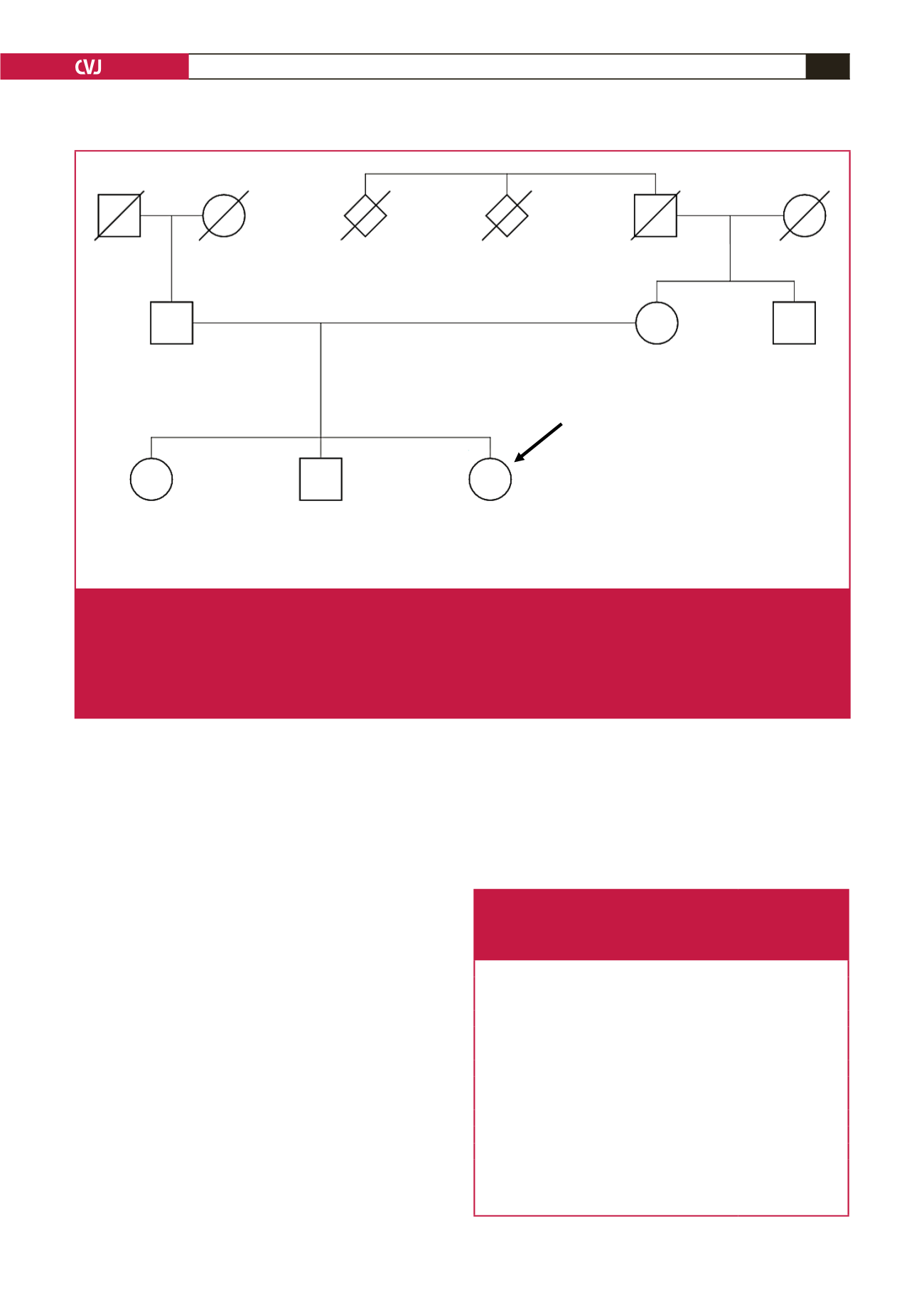
Fig. 1.
Pedigree of an Afrikaner family, with a clinical diagnosis of FH, subjected to whole-exome sequencing (WES), after exclusion
of the common three LDL receptor mutations (D154N, D206E, V408M) in the index case. The LDLR studies were combined
with APOE genotyping.
8
WES confirmed the presence of the low-penetrance APOE e-4 allele (rs429358) in the father and
sister, who shares APOB rs1367117 with the index case, as inherited from their mother. The sister tested positive for all four
GLGC risk alleles detectable by WES while the index case had only three and a somewhat milder LDL-C level. Subsequent
to the congress, the 12 SNP polygenic LDL-C genotype score was found to be higher in the index case (0.931) and sister
(1.002) compared to the mother (0.78) and father (0.684), as a result of the contribution of both parents.
Table 1.The commonest LDL receptor mutations in the FH phenotype
at a Cape Town lipid clinic.The Afrikaner LDL receptor defects
predominated and explain almost 80% of those with an identifiable
defect in this gene.Testing for mutations in three exons identified the
majority of the subjects with mutations.
Mutations
Number
Percent
D206E*
519
50.1
V408M
#
239
23.1
D154N*
63
6.1
D200G*
53
5.1
Del197*
45
4.3
G361V
$
32
3.1
C356Y
$
25
2.4
R329X
!
22
2.1
F382S
#
19
1.8
c.941-4G
>
A
!
19
1.8
E207K*
13
1.3
(…87 mutations)
Patients with successful genotype: 1 196.
Total with heterozygous FH phenotype: 2 200.
*Exon 4 (5),
!
exon 7 (2),
$
exon 8 (2),
#
exon 9 (2).