
















 54 / 82
54 / 82

CARDIOVASCULAR JOURNAL OF AFRICA • Volume 29, No 1, January/February 2018
52
AFRICA
10-year cardiovascular mortality rates vary between 10 and 15%,
with a worse prognosis for patients with severe MR.
11,12
Alterations in the global structure of the LV in response to
primary MR have been reviewed in detail previously.
9
Briefly,
MR results in increases in LA volume, a reduction in FSV
and an increase in left ventricular preload. By mechanisms
that are unclear but are discussed in more detail below, the LV
responds to the increased preload by eccentric hypertrophy, with
a serial increase in myocyte sarcomeres and myofibril slippage
(Fig. 1).
13-18
Eccentric hypertrophy normalises afterload, as estimated
by mean systolic stress, compared to patients with aortic
regurgitation, leading to a period of so-called ‘compensation’.
19
However, the hypertrophy that develops is actually insufficient
to fully compensate for the wall stress that develops. This is due
to inadequate protein synthesis triggered by MR compared to
pressure overload,
16,20
and progressive deterioration in myocardial
function.
21
There is no clear explanation for this phenomenon but it has
been proposed that the lower systolic load in the case of MR may
result in a reduced hypertrophic response at a time when there is
a marked demand for an increased stroke volume.
21
Altered
cytoskeletal changes, such as microtubular density, may also play
a role.
22
With time, in the face of inadequate hypertrophy and a
dilating LV, systolic wall stress increases [based on the Laplace
effect where wall stress (
σ
) is directly related to the pressure
within the ventricle and its radius (Pr), and inversely related to
the wall thickness (2h);
σ
=
Pr/2h] due to the increases in LV
dimensions and inadequate hypertrophy.
21,23,24
Chronic increases in wall stress are detrimental to the
myocardium, resulting in activation of a number of complex
inflammatory and apoptotic pathways, in a similar manner to
heart failure from other causes. Ultimately, there is myocyte loss
and sliding displacement of cardiomyocytes, or cell slippage,
caused by disruption of the myocardial extracellular matrix
(ECM)–integrin linkages.
7,25
Various lines of evidence point to time-dependent changes
in the up- and downregulation of remodelling pathways in
chronic primary MR.
26
This process is initiated by diastolic
mechanical stretch due to an increase in end-diastolic wall
stress, leading to an early increase in reactive oxygen species
(ROS) generation, inflammatory cytokine expression and
neurohormonal activation, with increases in angiotensin II and
catecholamine levels. Early in the remodelling process there is
interstitial collagen loss and cell slippage but with time there
is myocyte apoptosis and pathological ECM fibrosis.
27
Chronic
decompensated MR ensues, and the LV resembles end-stage
dilated cardiomyopathy.
MR causes mechanical stretch, which triggers
mechanoreceptors and activates signal-trans-
duction pathways
Myocardial mechanoreception is currently poorly understood.
28-30
There is no evidence that specialised mechanosensory cells exist
in the myocardium and the role of stretch-activated channels in
sensing stretch is debatable.
29
Two systems appear tobeparticularly
important in mechanoreception in the cardiomyocyte: the
collagen–integrin–cytoskeleton connections
25,31
and sarcomere-
related signalling.
30
The contraction–relaxation cycle of the myocyte depends on
coordinated interaction between the thin actin filament and the
thick myosin filament within the myocyte sarcomere.
32
Actin is
bound directly to the Z-disc while myosin is bound indirectly
to the Z-disc via the giant elastic protein, titin.
33,34
In the normal
heart, titin is responsible for restoring the stretched sarcomere
to its resting length following active contraction.
33,34
However,
another important role for titin is in mechanoreception and the
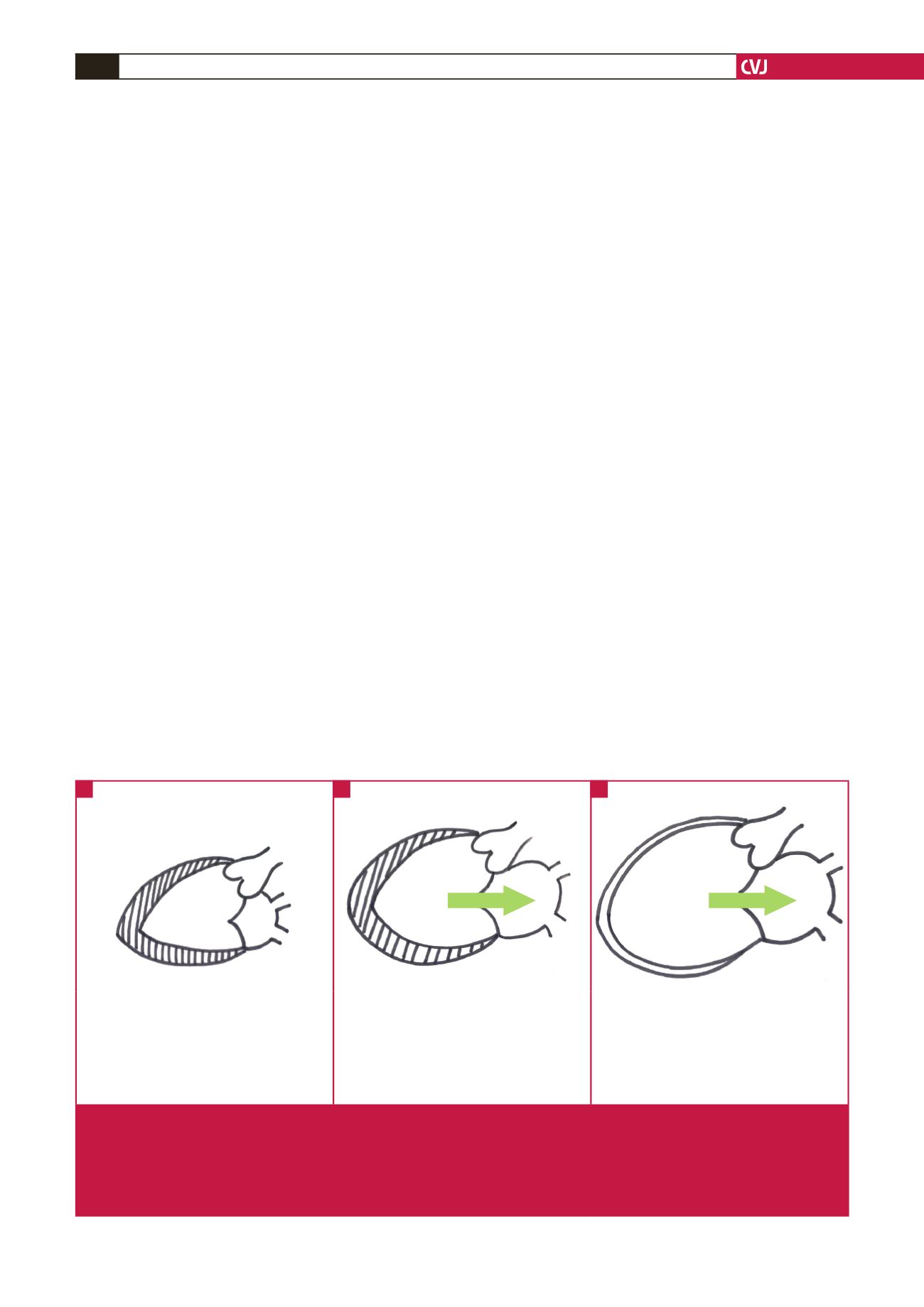
Normal heart
Chronic compensated MR
Decompensated MR
• Normal preload
• Normal LA volume
• Normal LV volume and contractility
• Normal wall stress
• Normal TSV and FSV
•
↑
preload
•
↑
LA volume and pressure
•
↑
LV volume
•
↑
contractility
• Eccentric hypertrophy but normalised wall
stress
•
↑
TSV
• Normal FSV
•
↑
preload
•
↑↑
LV volume
•
↓
contractility
•
↑↑
wall stress
•
↓
TSV
•
↓
FSV
Fig. 1.
Left ventricular remodelling in chronic primary mitral regurgitation. A: Normal LV is represented on the left. Wall stress is
normal. B: Chronic compensation with eccentric hypertrophy and dilatation. The increase in LV volume is compensated for
by the increase in wall thickness. Wall stress appears to be normalised by the eccentric hypertrophy. FSV is normal because
of increased LV filling. C: Adversely remodelled LV of decompensated chronic MR. The myocardial wall is thin resulting in an
increase in wall stress. The arrow indicates severe MR, which becomes more severe with a dilating LV. LA
=
left atrium; LV
=
left ventricle; TSV
=
total stroke volume; FSV
=
forward stroke volume; MR = mitral regurgitation.
A
B
C