


















 15 / 60
15 / 60

CARDIOVASCULAR JOURNAL OF AFRICA • Volume 32, No 1, January/February 2021
AFRICA
13
Norepinephrine and the retinal vasculature
In 2008, Krishnan and Nestler
36
discussed the monoamine
hypothesis of depression, which posits that depression is caused
by decreased monoamine function in the brain. Findings in the
low-u-NE group (tertile 1) support this low monoamine premise,
as initial low norepinephrine or monoamine concentrations
facilitated central neural control. An upregulation of
norepinephrine occurred over three years, resulting in neuronal
hyperactivity or adrenergic drive. In support, Ferrier
et al
.
6
suggested that the presence of chronic stress increases human
sympathetic firing, which is dependent on norepinephrine release
within the brain and the activation of central neural control
mechanisms to maintain homeostasis.
34,36
Catecholamine surges
following systemic insults, such as stress, is further directly
involved in the regulation of cytokine expression and exemplifies
the consistently high CRP level of 5 mg/l in the u-NE tertile 1, a
worsening clinical condition.
33-36
The noradrenergic cell groups A6 (locus cœruleus) project
axons to the hypothalamic paraventricular nucleus (PVN) to
activate the SAM and promote norepinephrine release during
acute and chronic stress.
11,36
However, the specific neurovascular
coupling mechanism in the human retina when neuronal
hyperactivity and chronic stress are apparent are not quite so
clear. Retinal neurons such as the amacrine and horizontal
cells synthesise catecholamines,
10
as well as high-affinity
α
1a
and
α
2a
-AR, which are expressed in Müller and ganglion cells, and
the inner plexiforme, inner nuclear and photoreceptor layers
of the retina.
10,22,37-39
Upon activation of
α
2a
-AR, norepinephrine
release is inhibited to protect ganglion cells against disturbed
ocular perfusion pressure
40
and arterial occlusion by reducing
intracellular cyclic adenosine monophosphate (cAMP)
production.
3,5
Müller cells and other astrocytes have intimate
contact with both synapses and blood vessels via
α
2a
-AR activity,
which enables regulation of blood flow.
41,42
A cycle of events may occur where chronic stress as the
initial trigger reflects low norepinephrine and potential
monoamine depletion (in u-NE tertile 1), and where central
homeostatic reflexes are activated to facilitate upregulation of
an endogenous catecholamine, norepinephrine. This happens
via activation of the hypothalamic PVN with sensitisation of
α
1a
-AR (vasoconstriction) and desensitisation of vasodilatory
α
2a
-AR in the retina. This may indicate that chronic stress
induces central neural control mechanisms, potentially over-
riding autoregulation in the retina. Furthermore, norepinephrine
may also bind with dopamine 2 receptors (D
2
R) as potential
signal transducers for norepinephrine in the outer and inner
retinal nuclear layers.
43
Retinal dopamine is synthesised and
released from dopaminergic amacrine cells
43
and binds to high-
affinity D
2
R to activate norepinephrine release.
43,44
In support
of this notion, Jäkel and Dimou
45
reported that hetero-receptor
cellular communication occurs between the different glial cell
types under pathological conditions.
Chronic stress may be such a pathological condition,
4
as
central neural control with upregulation of norepinephrine
in u-NE tertile 1 may have decreased
α
2a
-affinity/specificity.
This may allow D
2
R receptors to relay higher norepinephrine
vasoconstrictive signalling, thereby inducing reduced arterial
dilation, faster constriction, narrowing and hypo-perfusion in
u-NE tertile 1. Another study demonstrated desensitisation
of
α
1a
-AR upon provocation to protect the BRB,
10
potentially
explaining the lack of saliva
α
-amylase responses in our low-u-
NE cohort. This lack of variation in saliva
α
-amylase upon
provocation concurs with another study’s findings where chronic
stress or burn-out was associated with attenuated
α
-amylase
responses.
46
An adrenergic drive marker, depressed HRV, was also
suggested as an objective stress marker.
35
However, we could not
confirm HRV as a risk marker for either chronic cardiomyocyte
injury
25-27
or chronic stress and stroke risk in the current cohort.
The observed adrenergic drive or neuronal hyperactivity in
u-NE tertile 1 increased arterial vascular resistance and tone,
and may impair myogenic control. Considering the chronic
pre-diabetic status and adrenergic drive in u-NE tertile 1
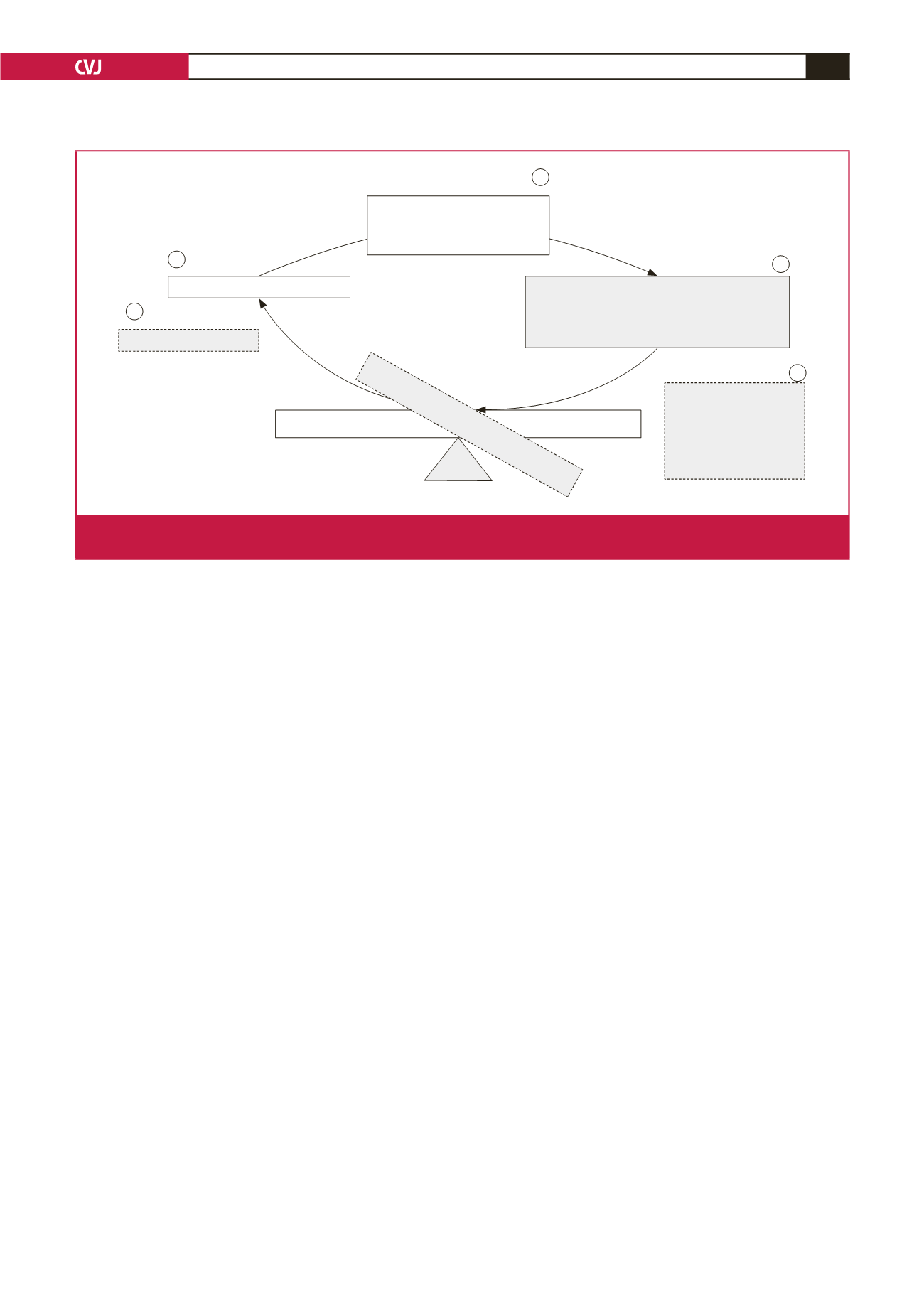
Low noreprinephrine
Retinal arterial narrowing
reflect
adrenergic drive
Retinal vein widening and delayed recovery
reflect
HPA axis hypo-secretion and
non-adaptation to stress
Chronic stress and
stroke risk:
•
↑
Arterial constriction
•
Hypo-perfusion
•
Delayed vein recovery
responses
•
Endothelial dysfunction
Taxing demands
Homeostasis
Blood-retinal barrier
CENTRAL NEURAL CONTROL
Facilitated
upregulation norepinephrine
dysregulation HPA axis
Breaching blood-retinal barrier
1
2
3
4
5
Fig. 4.
Graphical representation of the main findings indicating the relationship between chronic stress-related stroke risk and retinal
vein recovery responses.